Poster Presentations - Resistance to Molecular Targeted Therapies 3
Abstract 5649: Investigational study of acquired resistance to the EGFR irreversible inhibitor afatinib (BIBW2992) in wild-type and EGFR-mutant NSCLC cell lines.
Angela Alama, Simona Coco, Zita Cavalieri, Anna Truini, Cristina Bruzzo, Mariella Dono, and Francesco Grossi
IRCCS A.O.U. San Martino-IST, Genoa, Italy.
Introduction: Non-small cell lung cancer (NSCLC) represents about 85% of all lung cancers. Almost 20% of NSCLC tumors harbor somatic activating mutations of the tyrosine kinase (TK) encoding domain in EGFR gene (exons 18-21). Patients with EGFR mutations treated with specific inhibitors of EGFR-TKI such as gefitinib and erlotinib demonstrated a significantly longer survival. Nevertheless, the majority of patients acquire resistance to these drugs within one year due to a second-site mutation in EGFR (T790M). The irreversible EGFR-TKI, afatinib, has been found to be effective in inhibiting the growth of NSCLC cells with the T790M mutation of EGFR. In this study, we generated different NSCLC cell lines resistant to afatinib to investigate the biological and molecular mechanisms of acquired resistance.
Methods: A dose-escalation study to establish afatinib-resistant cell lines was performed in 3 NSCLC cell lines with a different EGFR mutational status: A549 (EGFR-wild type); H-1650 (exon19 delE746-A750); H-1975 (exon21 L858R/exon20 T790M). EGFR exons (18-21) from parental and resistant cells were sequenced by Sanger method while the EGF pathway was studied by qPCR using TaqMan® Array EGF Pathway and relative expression values of 92 genes, compared to parental cells, were obtained by 2–ddCT. Concomitantly, protein expression of some relevant genes such as EGFR and AKT, both phosphorylated and unphosphorylated, were investigated by western blot (WB).
Results: All 3 cell lines exhibited different degree of drug resistance and actively proliferated under persistent afatinib pressure (A549: 8.5μM; H-1650: 1.0μM; H-1975: 5.0μM) as compared to parental cultures. WB reported active phosphorylation of EGFR and AKT in resistant cells regardless of EGFR mutational status. This behavior also persisted in absence of EGF stimulation and in cultures maintained in afatinib-free medium for over 2 months. However none of the resistant cell lines harbored new EGFR mutation in the 4 exons (18-21). Gene expression analysis showed an increase of some members of RAS (MRAS, KRAS) and Rho families and also PI3K regulatory subunit 1 (PIK3R1) in A549 and H1975 cells. Notably, SHC3 (Src homology 2 domain containing; transforming protein 3) was up-regulated about 60-fold in H1975 whereas proto-oncogene JUN showed a 6-fold increase in H1650. In addition EGF was down-modulated in both H1650 and H1975.
Conclusion: Silencing of EGF in resistant cells together with the EGFR and AKT phosphorylation, suggest that these cultures acquired a different way to activate the EGF pathway. Furthermore in H-1975 the markedly increase of SHC3, an adaptor immediately downstream of EGFR, leads to hypothesize that it could be implicated in the activation of EGFR. Further investigations of SHC3 involvement in EGF pathway activation as well as sequencing analysis of the whole EGFR gene are ongoing.
|
 妈妈化疗的艰难时刻,倍瑞博营养液带
当母亲被确诊为肺腺癌四期的那一刻,我们全家如坠冰窟。随之而来的化疗更是煎熬——
妈妈化疗的艰难时刻,倍瑞博营养液带
当母亲被确诊为肺腺癌四期的那一刻,我们全家如坠冰窟。随之而来的化疗更是煎熬——
 希望给没有阅读过的人一些体会
本文来自于此书,只是做一些摘抄,分享给深陷迷茫的人。文中观点真实与否,效果
希望给没有阅读过的人一些体会
本文来自于此书,只是做一些摘抄,分享给深陷迷茫的人。文中观点真实与否,效果
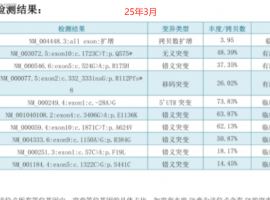 无靶点 her2扩增 二线白紫耐药后怎么
家父今年62岁,22年11月份颈部淋巴结穿刺,确诊肺低分化腺癌,分期是TxN3M1,基因检测
无靶点 her2扩增 二线白紫耐药后怎么
家父今年62岁,22年11月份颈部淋巴结穿刺,确诊肺低分化腺癌,分期是TxN3M1,基因检测
 父亲肺鳞癌,cT3N0M1a,ⅣA,脑转移
父亲2025年6月中旬因为头痛,一侧肢体不灵活入院,做了增强核磁,发现右侧额叶占位(3
父亲肺鳞癌,cT3N0M1a,ⅣA,脑转移
父亲2025年6月中旬因为头痛,一侧肢体不灵活入院,做了增强核磁,发现右侧额叶占位(3
 120万一针的抗癌药在肿瘤患者体内就
作者:Tony
一提起CAR-T,很多人都会想到的是其120万一针的恐怖价格,它是治愈癌症的
120万一针的抗癌药在肿瘤患者体内就
作者:Tony
一提起CAR-T,很多人都会想到的是其120万一针的恐怖价格,它是治愈癌症的





 显身卡
显身卡


